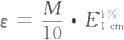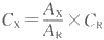|
|
|
÷–“©÷ΤΦΝΒΡΚ§ΝΩ≤βΕ®
ΖΔ≤Φ ±ΦδΘΚ2025/4/2 δ·άά¥Έ ΐΘΚ62
ΚΥ–ΡΡήΝΠ“Μ Ρή’ΐ»Ζ‘Υ”ΟΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»Ζ®Ϋχ––Κ§ΝΩ≤βΕ®
“Μ ΓΔΚΥ–ΡΗ≈Ρν 1.Ζ÷ΙβΙβΕ»Ζ® Ά®Ιΐ≤βΕ®±Μ≤βΈο÷ ‘ΎΧΊΕ®≤®≥Λ¥ΠΜρ“ΜΕ®≤®≥ΛΖΕΈßΡΎΒΡΈϋΙβΕ»ΜρΖΔΙβ«ΩΕ»Θ§Ε‘ΗΟΈο÷ Ϋχ––Ε®–‘ΜρΕ®ΝΩΖ÷ΈωΒΡΖΫΖ®ΓΘ‘ΎΩ…ΦϊΙβ«χΘ§≥ΐΡ≥–©Έο÷ Ε‘Ιβ”–Έϋ ’ΆβΘ§ΚήΕύΈο÷ ±Ψ…μ≤ΔΟΜ”–Έϋ ’Θ§ΒΪΩ…‘Ύ“ΜΕ®ΧθΦΰœ¬Φ”»κœ‘…ΪΦΝΜρΨ≠Ιΐ“ΜΕ®ΒΡ¥Πάμ ΙΤδœ‘…ΪΚσ‘Ό≤βΕ®Θ§Ι ”÷Ω…≥Τ±»…ΪΖ÷ΈωΓΘ 2.ΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»Ζ®(UV-Vis) “≤≥ΤΈΣΉœΆβ-Ω…ΦϊΈϋ ’ΙβΤΉΖ®Θ§ «―–ΨΩΈο÷ Ζ÷Ή”Ε‘ΉœΆβ-Ω…ΦϊΙβ(190ΓΪ800 nm) ΒΡΈϋ ’ΕχΫ®ΝΔΤπά¥ΒΡΖ÷ΈωΖΫΖ®ΓΘ ΕΰΓΔ―ßœΑΡΩ±ξ (1)Ρή λΝΖ Ι”ΟΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»Ζ®Ϋχ––÷–“©÷ΤΦΝΒΡΚ§ΝΩ≤βΕ®ΓΘ (2)Ρή λΝΖ Ι”ΟΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»ΦΤΓΘ »ΐ ΓΔΜυ±Ψ÷Σ Ε Ά®≥ΘΫΪ≤®≥Λ190ΓΪ400 nm ΖΕΈßΒΡΙβ≥ΤΈΣΉœΆβΙβΘΜ»Υ―έΡήΗ– ήΒΫΒΡΙβΒΡ≤®≥ΛΈΣ400ΓΪ800nm ΓΘΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»Ζ®ΨΆ «άϊ”ΟΈο÷ Ζ÷Ή”Έϋ ’190ΓΪ800 nm ΙβΤΉ«χΒΡΖχ…δά¥Ϋχ––Ζ÷Έω≤βΕ®ΒΡ“Μ÷÷ΖΫΖ®ΓΘ (“Μ)‘≠άμΘΚά ≤°-±»ΕϊΕ®¬…
ΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»Ζ®ΒΡΕ®ΝΩΖ÷Έω“άΨί «ά ≤°-±»ΕϊΕ®¬…Θ§ΤδΈοάμ“β“εΘΚΒ±“Μ χΤΫ––ΒΡΒΞ…ΪΙβΆ®ΙΐΨυ‘»ΓΔΖ«…Δ…δΧεœΒΒΡΒΆ≈®Ε»»ή“Κ ±Θ§‘ΎΒΞ…ΪΙβ«ΩΕ»ΓΔ»ή“ΚΈ¬Ε»Β»ΧθΦΰ≤Μ±δΒΡ«ιΩωœ¬Θ§ΈϋΙβΕ»”κΈϋ ’≥Ί(“Κ≤ψ)ΒΡΚώΕ»(Ιβ¬Ζ≥ΛΕ»)ΚΆΈϋΙβΈο÷ ΒΡ≈®Ε»ΒΡ≥ΥΜΐ≥…’ΐ±»ΓΘΤδ ΐ―ß±μ¥ο Ϋ»γœ¬ΘΚ ‘ΎΗχΕ®≤®≥ΛΓΔΫι÷ ΚΆΈ¬Ε»Β»ΧθΦΰœ¬Θ§Έϋ ’œΒ ΐ «Έο÷ ΒΡΧΊ–‘≥Θ ΐΘ§±μ ΨΈο÷ Ε‘Ρ≥“ΜΧΊΕ®≤®≥ΛΙβΒΡΈϋ ’ΡήΝΠΓΘ≤ΜΆ§Έο÷ Ε‘Ά§“Μ≤®≥ΛΒΡΒΞ…ΪΙβΩ…”–≤ΜΆ§ΒΡΈϋ ’œΒ ΐΓΘΈϋ ’œΒ ΐ‘Ϋ¥σΘ§±μ ΨΗΟΈο÷ Ε‘ΧΊΕ®≤®≥ΛΒΡΙβΒΡΈϋ ’ΡήΝΠ‘Ϋ«ΩΘ§≤βΕ®ΒΡΝιΟτΕ»‘ΫΗΏΘ§Υυ“‘Έϋ ’œΒ ΐ «Ε®–‘ΚΆΕ®ΝΩΒΡ“άΨίΓΘ Έϋ ’œΒ ΐΩ…Ζ÷ΈΣΑΌΖ÷Έϋ ’œΒ ΐΚΆΡΠΕϊΈϋ ’œΒ ΐΝΫ÷÷ΓΘΑΌΖ÷Έϋ ’œΒ ΐ”÷≥ΤΈΣ±»Έϋ ’œΒ ΐΘ§÷Η‘Ύ“ΜΕ®≤®≥Λœ¬Θ§»ή“Κ≈®Ε»ΈΣ1%(g/100 ml)ΓΔΈϋ ’≥ΊΚώΕ»ΈΣ1cm ±ΒΡΈϋΙβΕ»Θ§”ΟEl% ±μ ΨΘΜΡΠΕϊΈϋ ’œΒ ΐ÷Η‘Ύ“ΜΕ®≤®≥Λœ¬Θ§»ή“Κ≈®Ε»ΈΣ1 mol/L ΓΔ“Κ≤ψΚώΕ»ΈΣ1 cm ±ΒΡΈϋΙβΕ»Θ§”Οe±μ ΨΓΘ
ΝΫ÷÷Έϋ ’œΒ ΐ÷°ΦδΒΡΙΊœΒΘΚ (Εΰ)ΤΪάκά ≤°-±»ΕϊΕ®¬…ΒΡ“ρΥΊ “ΐΤπΤΪάκά ≤°-±»ΕϊΕ®¬…ΒΡ‘≠“ρ÷ς“Σ”–»ΐΗωΖΫΟφΘ§Αϋά®ά ≤°-±»ΕϊΕ®¬…±Ψ…μΒΡΨ÷œό–‘“ΐΤπΒΡ ΤΪάκΓΔΫι÷ ≤ΜΨυ‘»“ΐΤπΒΡΤΪάκΚΆ“«Τς“ΐΤπΒΡΤΪάκΓΘ ΥΡ ΓΔΡήΝΠ―ΒΝΖ (“Μ)≤ΌΉς”ΟΈο ≥Θ”ΟΒΡΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»ΦΤΆ®≥Θ”…Έε≤ΩΖ÷Ήι≥…ΘΚΙβ‘¥ΓΔΒΞ…ΪΤςΓΔΈϋ ’≥ΊΓΔΦλ≤βΤςΚΆ ΐΨί¥Πάμ œΒΆ≥(ΆΦ8-1,±μ8-1ΓΔ±μ8-2)ΓΘ
ΆΦ8-1 ΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»ΦΤΜυ±ΨΫαΙΙ Ψ“βΆΦ ±μ8-1 ΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»ΦΤΜυ±ΨΫαΙΙΦρΫι
±μ8-2ΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»ΦΤ≥ΘΦϊάύ–Ά
(Εΰ)ΉΔ“β ¬œν (1)Υυ”ΟΒΡΝΩΤΩΓΔ“Τ“ΚΙήΦΑΈϋ ’≥ΊΨυ”ΠΨ≠œ¥ΨΜΚσ Ι”ΟΓΘ (2)‘Ύ Ι”ΟΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»ΦΤ«ΑΘ§”ΠΕ‘Έϋ ’≥ΊΓΔΉν¥σΈϋ ’≤®≥ΛΓΔ‘”…ΔΙβΓΔœΝΖλΩμΕ»ΓΔΈϋΙβΕ»ΒΡΉΦ»ΖΕ»Β»Ϋχ–––Θ’ΐΚΆΦλΕ®Θ§ΖΫΖ®ΦϊΚ§ΝΩ≤βΕ®÷–ΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»ΦΤΒΡ–Θ’ΐœνœ¬ΓΘ (3)≤βΕ®÷ΤΦΝΚ§ΝΩ ±Θ§”Π ΙΙ© ‘ΤΖ»ή“Κ”κΕ‘’’ΤΖ»ή“ΚΨΏ”–œύΆ§ΒΡpH ÷ΒΓΘ (»ΐ)≤ΌΉςΡΎ»ί 1. Β―ι«ΑΉΦ±Η
2.ΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»ΦΤ Ι”Ο
≤ΜΆ§–ΆΚ≈ΒΡΉœΆβ-Ω…ΦϊΖ÷ΙβΙβΕ»ΦΤΒΡ≤ΌΉςΖΫΖ®ΚΆ“Σ«σ”–Υυ≤ΜΆ§Θ§ Ι”Ο«Α”ΠœξœΗ‘ΡΕΝ Ι”ΟΥΒ Ος ιΓΘ 3. ΐΨί¥Πάμ (1)ΒΞΉιΖ÷Ι© ‘ΤΖΒΡΕ®ΝΩΖ÷ΈωΓΘ
ΚΥ–ΡΡήΝΠΕΰ Ρή’ΐ»Ζ‘Υ”Ο±Γ≤ψ…®ΟηΖ®Ϋχ––Κ§ΝΩ≤βΕ®
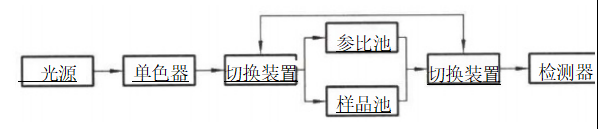
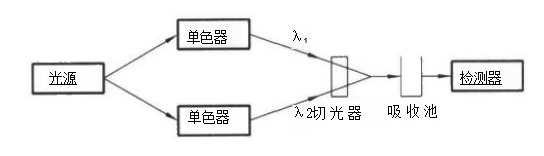
“Μ ΓΔΚΥ–ΡΗ≈Ρν ±Γ≤ψ…®ΟηΖ® Ι© ‘ΤΖΨ≠±Γ≤ψΖ÷άκΚσΘ§”Ο“Μ χ≤®≥ΛΓΔ«ΩΕ»“ΜΕ®ΒΡΉœΆβΙβΜρΩ…ΦϊΙβΕ‘±Γ≤ψΑεΫχ ––…®ΟηΘ§Ά®Ιΐ≤βΕ®±Γ≤ψΑε…œΒΡΑΏΒψΕ‘ΙβΒΡΈϋ ’«ΩΕ»ΜρΑΏΒψΨ≠ΦΛΖΔΚσΥυ≤ζ…ζΒΡ”ΪΙβ«ΩΕ»Θ§ΗυΨί…® ΟηΒΟΒΫΒΡΆΦΤΉΦΑΜΐΖ÷ ΐΨίΫχ––Ε®ΝΩΒΡΖΫΖ®ΓΘ Εΰ ΓΔ―ßœΑΡΩ±ξ (1)Ρή λΝΖ Ι”Ο±Γ≤ψ…®ΟηΖ®Ϋχ––÷–“©÷ΤΦΝΒΡΚ§ΝΩ≤βΕ®ΓΘ (2)Ρή λΝΖ≤ΌΉς±Γ≤ψ…®Οη“«ΓΘ »ΐ ΓΔΜυ±Ψ÷Σ Ε ±Γ≤ψ…®ΟηΖ®ΨΏ”–…η±ΗΦρΒΞΓΔ≤ΌΉςΖΫ±ψΓΔΖ÷άκΩλΥΌΓΔΝιΟτΕ»ΦΑΖ÷±φ¬ ΗΏΓΔœ‘…ΪΦΝ―Γ‘ώΖΕΈßΙψΓΔ ”Ο”ΎΕύΉιΖ÷ΦΑΈΔΝΩΉιΖ÷Ε®ΝΩΒ»”≈ΒψΘ§‘Ύ÷–“©÷ΤΦΝΦλ≤β÷–ΒΟΒΫΙψΖΚΒΡ”Π”ΟΓΘ»γΝυΈΕΒΊΜΤΆη÷ΤΦΝ÷– …Ϋήοί«ΓΔ…ΫιΪΜ·÷ΆΆη÷ΤΦΝ÷–…ΫιΪΒΡΚ§ΝΩ≤βΕ®Β»Ψυ≤…”ΟΥΪ≤®≥Λ…®ΟηΖ®Θ§ΒΦ≥ύΆη÷ΤΦΝ÷–ΜΤΝ§ΓΔΜΤΑΊΒΡΚ§ΝΩ≤βΕ®≤…”ΟΒΡ «±Γ≤ψ”ΪΙβ…®ΟηΖ®ΓΘ ±Γ≤ψ…®ΟηΖ®ΒΡ≤βΕ®‘≠άμ «”Ο≥ΛΩμΩ…“‘Βς’ϊΒΡ“Μ χ≤®≥ΛΓΔ«ΩΕ»ΙΧΕ®ΒΡΙβΘ§Ε‘±Γ≤ψΑε…œΒΡΑΏΒψΫχ––…®ΟηΘ§Ά®Ιΐ≤βΝΩΙβ χΨ≠ΙΐΑΏΒψΚσΒΡ«ΩΕ»±δΜ·Θ§«σ≥ω¥ΐ≤βΉιΖ÷Κ§ΝΩΓΘ±Γ≤ψ…®ΟηΖ®Ω…Ζ÷ΈΣ±Γ≤ψ Έϋ ’…®ΟηΖ®ΚΆ±Γ≤ψ”ΪΙβ…®ΟηΖ®ΓΘ 1.±Γ≤ψΈϋ ’…®ΟηΖ® ”ΟΩ…Φϊ-ΉœΆβΙβΒΡΒΞ…ΪΙβΕ‘±Γ≤ψΑε…œ…ΪΤΉΑΏΒψΫχ––…®ΟηΘ§ΗυΨί…®ΟηΜώΒΟΒΡΈϋΙβΕ»Υφ’ΙΩΣΨύάκ±δΜ·ΒΡΆΦΤΉΦΑΜΐΖ÷ ΐΨίΫχ––Ε®ΝΩΒΡΖΫΖ®ΓΘΗυΨί≤βΙβΒΡΖΫ Ϋ≤ΜΆ§Θ§±Γ≤ψΈϋ ’…®ΟηΖ®”÷Ζ÷ΈΣΆΗ…δΖ®ΚΆΖ¥…δΖ®ΝΫ÷÷ΓΘΆΗ…δΖ® «Ά®Ιΐ≤βΕ®ΙβΆΗΙΐΙ© ‘ΤΖΑΏΒψΚσΒΡΈϋ ’«ιΩωΫχ––Ε®ΝΩΒΡΖΫΖ®ΘΜΖ¥…δΖ® «Ά®Ιΐ≤βΕ®Ι© ‘ΤΖΑΏΒψΕ‘ΙβΒΡΖ¥…δ«ιΩωΫχ––Ε®ΝΩΒΡΖΫΖ®Θ§Τδ÷–“‘Ζ¥…δΖ®”Π”ΟΫœΈΣΤ’±ιΓΘ
”…”ΎΙβΒΡΈϋ ’Ε®¬…“Σ«σΙβΆ®ΙΐΒΡΙ© ‘ΤΖ «Ψυ‘»ΓΔΈό…Δ…δΒΡΫι÷ Θ§Εχ±Γ≤ψΑε «”…–μΕύœΗ–ΓΒΡΈϋΗΫΦΝΩ≈ΝΘΉι≥…ΒΡΑκΆΗΟςΈοΧεΘ§Β±Ιβ’’…δΒΫ±Γ≤ψΑε±μΟφ ±Θ§Ιβ≥ΐ±ΜΈϋ ’“‘ΆβΘ§ΜΙΜα≤ζ…ζΆΗ…δΓΔΖ¥…δΦΑ…Δ…δΒ»œ÷œσΘ§ ΙΙ© ‘ΤΖΒΡΈϋΙβΕ»”κΈο÷ ΒΡ≈®Ε»ΦΑ“Κ≤ψΚώΕ»≤Μ≥ œΏ–‘ΙΊœΒΘ§ΗχΕ®ΝΩΖ÷ΈωΙΛΉς¥χά¥ΝΥάßΡ―Θ§ΈΣ¥ΥΩ…≤…”Ο“‘œ¬»ΐ÷÷ΖΫΖ®ΫβΨωΓΘ
(1)―Γ‘ώ«ζœΏ÷–ΒΡ÷±œΏ≤ΩΖ÷Ϋχ––Ε®ΝΩΖ÷ΈωΘΚΩ…άϊ”Ο‘ΎΒΆ≈®Ε»ΖΕΈßΡΎΘ§±Γ≤ψΑε…œΑΏΒψΒΡΙ© ‘ΤΖΝΩ”κ≤βΒΟ÷Β≥ œΏ–‘ΙΊœΒΫχ––Ε®ΝΩΖ÷ΈωΓΘΒΪœΏ–‘ΖΕΈßΫœ’≠Θ§”Π”Ο…œ ήΒΫ“ΜΕ®œό÷ΤΓΘ¥ΥΖ®Φρ±ψΘ§ «ΟΜ”–œΏ–‘Μ·ΙΠΡήΒΡ“«ΤςΉν≥Θ Ι”ΟΒΡΖΫΖ®ΓΘ
(2)≤…”ΟΩβ±¥ΕϊΩ®-Ο…ΩΥΖΫ≥ΧΘΚΩβ±¥ΕϊΩ®ΦΑΟ…ΩΥΕ‘Ιβ’’ΒΫ±Γ≤ψΑε…œΒΡ––ΈΣΫχ––Ζ÷Έω Β―ιΘ§¥”άμ¬έ…œΆΤΒΦ≥ωΦρΜ·ΒΡΖΫ≥Χ ΫΘ§”Π”ΟΒΫ…®Οη“«…œΓΘΒ±Ιβ’’…δΒΫ±Γ≤ψΑε…œΒΡΑΏΒψ ±Θ§Ιβ≥ΐΝΥ±ΜΙ© ‘ΤΖΈϋ ’ΓΔΖ¥…δΓΔ…Δ…δΓΔΆΗ…δΆβΘ§ΜΙ±ΜΩ’ΑΉ±Γ≤ψΑε(ΈϋΗΫΦΝΓΔπΛΚœΦΝΓΔ≤ΘΝßΑεΒ»)Έϋ ’ΓΔΖ¥…δΓΔ…Δ…δΓΔΆΗ…δΓΘ‘Ύ ΒΦ ≤βΝΩ ±Θ§ΈΣΝΥœϊ≥ΐΩ’ΑΉ±Γ≤ψΑεΕ‘≤βΕ®ΫαΙϊΒΡ”ΑœλΘ§Ά®≥Θœ»Ε‘Ω’ΑΉ±Γ≤ψΑεΫχ––…®ΟηΘ§ΫΪ Ω’ΑΉ±Γ≤ψΑεΒΡ”Αœλ÷ΒΉςΈΣΜυΉΦΘ§≤βΕ®ΑΏΒψΒΡœύΕ‘ΆΗΙβ¬ ΜρœύΕ‘Ζ¥…δ¬ Θ§ΦΤΥψ±Γ≤ψΑε…œΑΏΒψΒΡΈϋ ’Ε»ΦΑΚ§ΝΩΓΘ
‘Ύ“Μ–©“«Τς÷–”–œΏ–‘Μ·ΤςΘ§Ω…ΗυΨίΩβ±¥ΕϊΩ®-Ο…ΩΥΖΫ≥ΧΫΪ«ζœΏ–Θ’ΐΈΣ÷±œΏΘ§”Ο–Θ’ΐΚσΒΡ÷±œΏΕ®ΝΩ ΓΘ
(3)άϊ”ΟΖ«œΏ–‘ΖΫ≥ΧΕ®ΝΩΘΚάϊ”ΟΦΤΥψΜζœ»«σ≥ωΖ«œΏ–‘ΖΫ≥ΧΘ§»ΜΚσΫΪΙ© ‘ΤΖΒΡ≤βΕ®÷Β δ»κΦΤΥψΜζΘ§”…¥ΥΖΫ≥Χ«σ≥ωΙ© ‘ΤΖΒΡ≈®Ε»ΓΘ
2.±Γ≤ψ”ΪΙβ…®ΟηΖ® Ά®Ιΐ“«ΤςΕ‘Ι© ‘ΤΖΈϋ ’Ω…Φϊ-ΉœΆβΙβΚσΖΔ…δΒΡ”ΪΙβ«ΩΕ»Ϋχ––…®Οη“‘ΒΟΒΫΈϋ ’ΙβΤΉΦΑΜΐΖ÷ ΐΨίΘ§¥”Εχ«σ≥ωΙ© ‘ΤΖΚ§ΝΩΒΡΖΫΖ®ΓΘΤδΝιΟτΕ»±»±Γ≤ψΈϋ ’…®ΟηΖ®ΗΏ1~3Ηω ΐΝΩΦΕΘ§ΒΪ ”ΟΖΕΈßΫœ’≠Θ§Ω…Ά®Ιΐ”ΪΙβ―ή…ζΜ·Ζ¥”Πά©¥σ”Π”ΟΖΕΈßΓΘ
‘Ύ±Γ≤ψ”ΪΙβ…®ΟηΖ®÷–Θ§Β±ΒψΦ”ΒΡ―υΤΖΝΩΚή…Ό ±Θ§ΑΏΒψ÷–ΉιΖ÷ΒΡ≈®Ε»”κ”ΪΙβ«ΩΕ»≥ œΏ–‘ΙΊœΒΘ§Ω…Α¥œ¬ ωΙΪ ΫΦΤΥψΘΚ
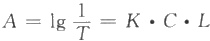
F=2.3K/I₀EΓΛCΓΛL Μρ F=K'ΓΛC
Ϋ÷–Θ§F ΈΣ”ΪΙβΈο÷ ΒΡ”ΪΙβ«ΩΕ»ΘΜIΓΘΈΣ»κ…δΙβ«ΩΕ»ΘΜCΈΣ¥ΐ≤βΉιΖ÷ΒΡ≈®Ε»ΘΜE ΈΣΈϋ ’œΒ ΐΘΜKΚΆK'ΈΣ≥Θ ΐ(”κ”ΪΙβ–߬ ”–ΙΊ) ;L ΈΣ±Γ≤ψΚώΕ»ΘΜK'=2.3K/IΓΘΓΛEΓΛLΓΘ
‘Ύ±Γ≤ψ”ΪΙβ…®ΟηΖ®÷–Θ§≥Θ”ΟΑΏΒψ”ΪΙβ«ΩΕ»ΒΡΜΐΖ÷÷Β(…ΪΤΉΖεΟφΜΐ)¥ζΧφΙΪ Ϋ÷–ΒΡF,ΑΏΒψ÷–ΉιΖ÷ΒΡΚ§ΝΩ¥ζΧφΙΪ Ϋ÷–ΒΡCΫχ––ΦΤΥψΓΘ
ΥΡΓΔΡήΝΠ―ΒΝΖ
(“Μ)≤ΌΉς”ΟΈο
–ρΚ≈
“«ΤςΉι≥…
Φρ Ϋι
1
Ιβ‘¥
Ιβ‘¥ «ΡήΧαΙ©Έ»Ε®ΒΡΓΔΨΏ”–“ΜΕ®«ΩΕ»ΒΡΥυ–η≤®≥ΛΖΕΈßΡΎΒΡΝ§–χΙβ‘¥Θ§Αϋά®ΉœΆβΙβ‘¥κ°ΒΤ(200ΓΪ370 nm);Ω…ΦϊΙβ‘¥ΈΌΒΤ(370ΓΪ700 nm);”ΪΙβΙβ‘¥κ·ΒΤΜρΙ·ΒΤ(200~
700 nm),Τδ÷–Ι·ΒΤΈΣœΏΙβ‘¥Θ§Ω…ΧαΙ©ΧΊ’ςΖχ…δΙβΤΉ
2
ΒΞ…ΪΤς
ΒΞ…ΪΤςΩ…“‘ΫΪΙβ‘¥ΖΔ≥ωΒΡΝ§–χΙβΖ÷ΫβΈΣΒΞ…ΪΙβΓΘΤδ÷ς“Σ”…»κ…δœΝΖλΓΔ≥ω…δœΝΖλΓΔ…Ϊ…Δ‘ΣΦΰΓΔΤΫ––ΙβΉΑ÷Ο(ΉΦ÷±ΨΒ)Β»≤ΩΖ÷ΙΙ≥…ΓΘ±Γ≤ψ…®Οη“«Εύ≤…”ΟΙβ’ΛΈΣ…Ϊ…Δ‘ΣΦΰ
3
±Γ≤ψΑεΧ®
±Γ≤ψΑεΧ®Αϋά®±Γ≤ψΑεΧ®ΦήΦΑ«ΐΕ·ΉΑ÷ΟΓΘ…®Οη ±±Γ≤ψΑεΙΧΕ®”Ύ±Γ≤ψΑεΧ®Φή…œΘ§±Γ≤ψΑεΧ®ΦήΩ…Ϋχ––ΚαœρΓΔΉίœρ“ΤΕ·Θ§ Ι±Γ≤ψΑεΉωX÷αΚΆY÷αΖΫœρΒΡ“ΤΕ·Θ§Άξ≥…Ε‘Ι© ‘ΤΖΑΏΒψΒΡΆΗ…δΙβΓΔΖ¥…δΙβΦΑΖΔ≥ω”ΪΙβΒΡ…®ΟηΙΛΉς
4
Φλ≤βΤς
Φλ≤βΤς «”Οά¥Φλ≤βΚΆΖ≈¥σ”…Ι© ‘ΤΖΑΏΒψΆΗ…δΓΔΖ¥…δΜρΖΔ≥ω”ΪΙβ«ΩΕ»ΒΡΉΑ÷ΟΓΘ
±Γ≤ψ…®Οη“«ΒΡΦλ≤βΤςΕύΈΣΙβΒγ±Ε‘ωΙήΓΘ÷ς“Σ”–Φλ≤β»κ…δΙβ”ΟΒΡ≤Έ±»ΙβΒγ±Ε‘ωΙήΓΔΦλ≤β”ΪΙβ”ΟΒΡΙβΒγ±Ε‘ωΙήΓΔΦλ≤βΖ¥…δΙβ”ΟΒΡΙβΒγ±Ε‘ωΙήΦΑΦλ≤βΆΗ…δΙβ”ΟΒΡΙβΒγ±Ε‘ωΙήΓΘΙβΒγ±Ε‘ωΙήΆ®ΙΐΫΪΙβ–≈Κ≈ΉΣ±δΈΣΒγ–≈Κ≈ΚσΘ§Ά®Ιΐ“ΜœΒΝ–ΒΡ±Ε‘ωΙΐ≥ΧΫΪ’β–©Βγ–≈Κ≈Ζ≈¥σΘ§¥”Εχ Βœ÷Ε‘Ιβ«ΩΕ»ΒΡΦλ≤βΓΘ
Φλ≤βΖ¥…δΙβ ±Θ§Ιβ±ΜΖ÷≥…ΝΫ χ«ΩΕ»œύΒ»ΒΡΙβΘ§“Μ χΙβ”… ·”Δ¥ΑΑεΖ¥…δΒΫΦλ≤β”ΟΙβΒγ±Ε‘ωΙήΘΜΝμ“Μ χΙβ’’…δΒΫΙ© ‘ΤΖΑΏΒψ…œΘ§“Μ≤ΩΖ÷±ΜΙ© ‘ΤΖΈϋ ’Θ§Νμ“Μ≤ΩΖ÷ΖΔ…ζ…Δ…δΘ§…Δ…δΙβ±ΜΖ¥…δ”ΟΙβΒγ±Ε‘ωΙήΥυΫ” ’ΓΘΦλ≤β”ΟΙβΒγ±Ε‘ωΙή”κΖ¥…δ”ΟΙβΒγ±Ε‘ωΙή δ≥ω–≈Κ≈÷°±»Θ§Ψ≠Ε‘ ΐΉΣΜΜΤςΉΣΜΜΈΣΈϋ ’Ε»–≈Κ≈ΓΘ
Φλ≤βΆΗ…δΙβ ±Θ§”…ΆΗ…δ”ΟΙβΒγ±Ε‘ωΙή¥ζΧφΖ¥…δ”ΟΙβΒγ±Ε‘ωΙήΓΘΥϋΒΡ δ≥ω–≈Κ≈”κΦλ≤β”ΟΙβΒγ±Ε‘ωΙήΒΡ δ≥ω–≈Κ≈÷°±»Θ§Ψ≠Ε‘ ΐΉΣΜΜΤςΉΣΜΜΚσΒΟΒΫΆΗ…δ≤βΕ®ΒΡΈϋ ’Ε»–≈Κ≈
5
ΙΛΉς’Ψ
ΨΏ”–…ηΕ®“«Τς≤Έ ΐΓΔΩΊ÷Τ“«ΤςΙΛΉςΧθΦΰΚΆ–≈Κ≈≤…Φ·ΓΔ¥ΠάμΦΤΥψΚΆ¥ρ”Γ δ≥ωΒ»ΙΠΡή
(Εΰ)ΉΔ“β ¬œν
(1)≥ΐΝμ”–ΙφΕ®ΆβΘ§±Γ≤ψ…®ΟηΖ®Κ§ΝΩ≤βΕ®”Π Ι”Ο – έ±Γ≤ψΑεΓΘ
(2)±Γ≤ψ…ΪΤΉΖ÷ΈωΒΡΗςΗω≤Ϋ÷η»γΒψ―υΓΔ’ΙΩΣΒ»Θ§ΨυΜα”Αœλ±Γ≤ψ…®ΟηΫαΙϊΒΡΉΦ»Ζ–‘”κ÷Ίœ÷–‘Θ§“ρ¥Υ‘ΎΦλ≤βΒΡΗςΗω≤Ϋ÷η”ΠΙφΖΕ≤ΌΉςΓΘ
(3)…®Οη ±”Π―Ί’ΙΩΣΖΫœρΉ‘œ¬Εχ…œΫχ––…®ΟηΘ§≤ΜΡήΚαœρ…®ΟηΓΘ
(4)≤βΕ®Φ«¬Φ÷–”ΠΑϋΚ§±Γ≤ψ…®ΟηΆΦΓΔΖεΟφΜΐΜΐΖ÷÷ΒΓΔΙΛΉς«ζœΏΓΔΜΊΙιΖΫ≥ΧΚΆœύΙΊœΒ ΐΦΑ≤βΕ®ΫαΙϊΦΤΥψΒ»ΓΘ
(5)ΗυΨί ΒΦ «ιΩωΘ§Ω…Βς’ϊΙ© ‘ΤΖ»ή“ΚΦΑΕ‘’’ΤΖ»ή“ΚΒΡΒψ―υΝΩΘ§“‘±ψ”Ύ≤βΕ®ΓΘ
(6)ΈΣ±Θ÷Λ≤βΕ®ΫαΙϊΒΡΉΦ»Ζ–‘Θ§≤…”ΟΆβ±ξ“ΜΒψΖ®≤βΕ® ±Θ§Ι© ‘ΤΖΑΏΒψ”Π”κΕ‘’’ΤΖΑΏΒψΒΡΖεΟφΜΐΒΡ÷ΒΫ”ΫϋΘΜ≤…”ΟΆβ±ξΝΫΒψΖ®≤βΕ® ±Θ§Ι© ‘ΤΖΑΏΒψΒΡΖεΟφΜΐ”Π‘ΎΝΫΗωΕ‘’’ΤΖΑΏΒψΒΡΖεΟφΜΐ÷Β÷°Φδ ΓΘ
(»ΐ)≤ΌΉςΡΎ»ί
–ρΚ≈
≤ΌΉς≤Ϋ÷η
ΦΦ Ρή “Σ «σ
1
≤β ‘»ή“Κ ΒΡ÷Τ±Η
ΫΪΕ‘’’ΤΖΦΑΙ© ‘ΤΖΑ¥ΓΕ÷–Ιζ“©ΒδΓΖ±ξΉΦ÷–Ηςœνœ¬ΒΡΙφΕ®Ϋχ––≈δ÷ΤΘ§“ΜΑψ”Π»Γ2Ζί»ή “ΚΫχ––ΤΫ––≤ΌΉςΓΘ”Π”ΟΆβ±ξ“ΜΒψΖ® ±÷Μ–η“Μ÷÷≈®Ε»ΒΡΕ‘’’ΤΖΦΑΙ© ‘ΤΖ»ή“ΚΘ§”Π”ΟΆβ ±ξΝΫΒψΖ® ±Ω…≈δ÷Τ“Μ÷÷ΜρΝΫ÷÷≈®Ε»Ε‘’’ΤΖ»ή“Κ
2
±Γ≤ψ…ΪΤΉ ≤ΌΉς
(1)Άβ±ξ“ΜΒψΖ®ΘΚ”ΟΒψ―υΤςΖ÷±πΫΪ“Μ÷÷≈®Ε»ΒΡΙ© ‘ΤΖ»ή“Κ”κΕ‘’’ΤΖ»ή“ΚΫΜ≤φΒψ”ΎΆ§ “Μ±Γ≤ψΑε…œΘ§‘≠Βψ÷±ΨΕ2 mmΉσ”“Θ§Ι© ‘ΤΖ”κΕ‘’’ΤΖΒΡΑΏΒψ ΐΡΩ≤ΜΒΟ…Ό”Ύ4ΗωΘ§ΟΩΗωΑΏ ΒψΒΡΒψ―υΝΩ“ΣœύΆ§
(2)Άβ±ξΝΫΒψΖ®ΘΚ”ΟΒψ―υΤςΖ÷±πΫΪ“Μ÷÷≈®Ε»ΒΡΙ© ‘ΤΖ»ή“Κ”κΝΫ÷÷≈®Ε»Μρ÷ΊΝΩΒΡΕ‘’’ ΤΖ»ή“ΚΫΜ≤φΒψ”ΎΆ§“Μ±Γ≤ψΑε…œΘ§‘≠Βψ÷±ΨΕ2 mmΉσ”“ΓΘΙ© ‘ΤΖΒΡΑΏΒψ≤ΜΒΟ…Ό”Ύ4ΗωΘ§
Ά§“Μ≈®Ε»Μρ÷ ΝΩΒΡΕ‘’’ΤΖΑΏΒψ≤ΜΒΟ…Ό”Ύ2ΗωΓΘΙ© ‘ΤΖΟΩΗωΑΏΒψΒΡΒψ―υΝΩ“ΣœύΆ§Θ§Ά§ “Μ≈®Ε»Ε‘’’ΤΖΒΡΒψ―υΝΩ“ΣœύΆ§
3
’ΙΩΣ
≥ΐΝμ”–ΙφΕ®ΆβΘ§Α¥“©ΤΖ±ξΉΦ÷–ΗςΤΖ÷÷œνœ¬ΒΡΨΏΧεΙφΕ®’ΙΩΣ≤ΌΉς
4
œΒΆ≥ ”Ο –‘ ‘―ι
(1)ΝιΟτΕ»ΘΚœόΝΩΦλ≤ι ±Θ§±Μ≤βΉιΖ÷Ρή±ΜΦλ≥ωΒΡΉνΒΆΝΩΓΘ“ΜΑψ≤…”ΟΕ‘’’ΤΖ»ή“Κ”κœΓ Ά»τΗ…±ΕΒΡΕ‘’’ΤΖ»ή“Κ‘ΎΙφΕ®…ΪΤΉΧθΦΰœ¬Θ§‘ΎΆ§“Μ±Γ≤ψΑε…œΒψ―υΓΔ’ΙΩΣΚσΫχ––Φλ ”Θ§œ‘«εΈζΒΡΑΏΒψΒΡΉνΒΆ≈®Ε»»ή“ΚΒΡΒψ―υΝΩΦ¥ΈΣΝιΟτΕ»
(2)Ζ÷άκΕ»ΘΚ”Ο”ΎΦχ±π ±Θ§Ε‘’’ΤΖ»ή“Κ”κΙ© ‘ΤΖ»ή“Κ…ΪΤΉ÷–œύ”ΠΒΡ÷ςΑΏΒψΘ§Ψυ”Πœ‘ ΨΝΫΗω«εΈζΖ÷άκΒΡΑΏΒψΓΘ”Ο”ΎœόΝΩΦλ≤ιΜρΚ§ΝΩ≤βΕ® ±Θ§“Σ«σΕ®ΝΩΖε”κœύΝΎΖε÷°Φδ”–ΫœΚΟΒΡΖ÷άκΕ»ΓΘΖ÷άκΕ»(R)ΒΡΦΤΥψΙΪ ΫΈΣ
R=2(d₁-d₂)/W₁+W₂
Ϋ÷–ΘΚd₁ΈΣœύΝΎΝΫΖε÷–«Α“ΜΖε”κ‘≠ΒψΒΡΨύάκΘΜd₂ΈΣœύΝΎΝΫΖε÷–Κσ“ΜΖε”κ‘≠ΒψΒΡΨύάκΘΜW₁ΓΔW₂ΈΣœύΝΎΝΫΖεΗςΉ‘ΒΡΖεΩμΓΘ
≥ΐΝμ”–ΙφΕ®ΆβΘ§Ζ÷άκΕ»”Π¥σ”Ύ1.0
(3)÷ΊΗ¥–‘ΘΚΆ§“Μ±Γ≤ψΑε…œœύΆ§≈®Ε»ΒΡΆ§“ΜΙ© ‘ΤΖ»ή“Κ ΐΗωΑΏΒψΘ§…®ΟηΫαΙϊΒΡΤΪ≤νΓΘ»γ±Γ≤ψΑε’ΙΩΣΚσ÷±Ϋ”…®ΟηΘ§Ά§“Μ±Γ≤ψΑε…œΤΫ––Βψ―υΒΡ¥ΐ≤βΉιΖ÷ΑΏΒψ(≤Μ…Ό”Ύ4ΗωΒψ)
ΒΡΖεΟφΜΐ≤βΝΩ÷ΒΒΡœύΕ‘±ξΉΦΤΪ≤ν≤Μ”Π¥σ”Ύ3.0%;»γ–ηœ‘…ΪΚσ…®ΟηΘ§ΤδœύΕ‘±ξΉΦΤΪ≤ν≤Μ”Π¥σ”Ύ5.0%
5
…œΜζ…®Οη
(1)―Γ‘ώΦλ≤βΖΫΖ®ΘΚΦλ≤βΖΫΖ®”–±Γ≤ψΈϋ ’…®ΟηΖ®ΚΆ±Γ≤ψ”ΪΙβ…®ΟηΖ®ΓΘ‘ΎΉœΆβΜρΩ…ΦϊΙβ«χ”–Έϋ ’ΒΡΉιΖ÷Ω…―ÔϱΓ≤ψΈϋ ’…®ΟηΖ®Θ§”–”ΪΙβΜρΨ≠ Β±¥ΠάμΚσΩ…–Έ≥…”ΪΙβΒΡΉιΖ÷Ω…―Γ‘ώ±Γ≤ψ”ΪΙβ…®ΟηΖ®
(2)―Γ‘ώ≤βΕ®ΖΫ ΫΘΚ±Γ≤ψΈϋ ’…®ΟηΖ®ΒΡ≤βΙβΖΫ Ϋ”–Ζ¥…δΖ®ΚΆΆΗ…δΖ®ΓΘΖ¥…δΖ® «ΫΪΙβ χ’’…δΒΫ±Γ≤ψΑε…œΘ§≤βΝΩΖ¥…δΙβΒΡ«ΩΕ»ΘΜΆΗ…δΖ® «≤βΝΩΆΗ…δΙβΒΡ«ΩΕ»ΓΘ”…”Ύ±Γ≤ψ≤ΡΝœΕύΈΣΑΉ…ΪΙΧΧεΘ§Ε‘ΙβΒΡΆ®ΆΗ–‘≤ΜΚΟΘ§≤βΕ®ΫαΙϊΒΡΈσ≤ν¥σΘ§ΝμΆβΘ§≤ΘΝßΑεΕ‘–Γ”Ύ330nm≤®≥ΛΒΡΉœΆβΙβ”–Έϋ ’Θ§Υυ“‘≤ΜΡή”ΟΆΗ…δΖ®≤βΕ®Ε‘330nm≤®≥Λ“‘œ¬”–Έϋ ’ΒΡΈο÷ ΓΘ”… ”ΎΖ¥…δΖ®≤βΝΩΒΡ «Ζ¥…δΙβΘ§≤ΘΝßΑεΦΑ±Γ≤ψΚώΕ»ΒΡΈΔ–Γ±δΜ·Ε‘≤βΕ®ΫαΙϊ”Αœλ≤Μ¥σΘ§Υυ“‘‘ΎΕ®ΝΩΖ÷Έω÷–Εύ≤…”ΟΖ¥…δΖ®
(3)―Γ‘ώ…®Οη≤®≥ΛΘΚ±Γ≤ψ…®ΟηΩ… Ι”ΟΒΞ≤®≥ΛΚΆΥΪ≤®≥ΛΫχ––≤βΕ®ΓΘ
ΔΌΒΞ≤®≥Λ…®ΟηΖ®ΘΚ”Ο“Μ÷÷≤®≥ΛΒΡΙβœΏΕ‘±Γ≤ψΫχ––…®ΟηΘ§“ΜΑψ―Γ‘ώΠΥmaxΈΣ’’…δ≤®≥ΛΘ§ΗυΨί…®Οη ±ΙβΒΡ–Έ Ϋ≤ΜΆ§Θ§”÷Ζ÷≥…ΒΞΙβ χΦΑΥΪΙβ χΝΫ÷÷–Έ ΫΓΘΤδ÷–ΒΞ≤®≥ΛΓΔΒΞΙβ χ…®Οη Ε‘”Ύ±Γ≤ψΑεΚώΕ»≤ΜΨυ‘»ΓΔœ‘…Ϊ≤ΜΨυ‘»ΜρΤδΥϊ“ρΥΊ‘λ≥…ΒΡ±≥ΨΑ≤ΜΨυ‘»Β»”ΑœλΈόΖ®œϊ≥ΐΓΘ “ρ¥ΥΕ‘±Γ≤ψΑεΒΡ“Σ«σΫœΗΏΘ§ ”Ο”Ύ…®ΟηΜυœΏΤΫΈ»ΓΔΈό±≥ΨΑΗ…»≈ΒΡ±Γ≤ψΒΡ≤βΕ®ΓΘ
ΔΎΥΪ≤®≥Λ…®ΟηΖ®ΘΚ≤…”ΟΝΫ χ≤ΜΆ§≤®≥ΛΒΡΒΞ…ΪΙβΘ§œ»Κσ…®ΟηΥυ“Σ≤βΕ®ΒΡΑΏΒψΘ§“‘ΑΏΒψΕ‘ΝΫ χ≤ΜΆ§ΙβΒΡΈϋ ’Ε»≤ν÷ΒΫχ––Ε®ΝΩΓΘΩ…Ζ÷ΈΣΥΪ≤®≥ΛΓΔΥΪΙβ χ…®ΟηΖ®ΚΆΥΪ≤®≥ΛΓΔΒΞΙβ χ…®ΟηΖ®ΓΘ“ΜΑψ―Γ‘ώ±Μ…®ΟηΑΏΒψΒΡΉν¥σΈϋ ’≤®≥ΛΈΣ≤βΕ®≤®≥Λ(ΠΥs),”ΟΗΟΑΏΒψΈόΈϋ ’ΜρΉν–ΓΈϋ ’ΒΡ≤®≥ΛΈΣ≤Έ±»≤®≥Λ(ΠΥR)ΓΘ¥ΥΖ®Ω…œϊ≥ΐ±≥ΨΑΗ…»≈Θ§Φθ…ΌΖ÷άκΕ»«ΖΦ―ΒΡΝΫΗωΑΏΒψ÷°ΦδΒΡœύΜΞΗ…»≈Θ§…®ΟηΜυœΏΫœΈΣΤΫΈ»Θ§≤βΕ®ΨΪΟήΕ»ΒΟΒΫΗΡ…ΤΓΘ ”Ο”Ύ±≥ΨΑ≤ΜΨυ‘»ΒΡ±Γ≤ψΑεΒΡ…®ΟηΓΘ
Δέ±Γ≤ψ”ΪΙβ…®Οη≤®≥ΛΒΡ―Γ‘ώΘΚ”…”Ύ±Γ≤ψ”ΪΙβ…®Οη÷–ΑΏΒψΒΡ”ΪΙβΜΐΖ÷÷Β”κΉιΖ÷ΒΡΚ§ΝΩ≥ œΏ–‘ΙΊœΒΘ§“ρ¥ΥΘ§”ΠΙΊ±’œΏ–‘Μ·ΤςΓΘ±Γ≤ψ”ΪΙβ…®Οη ±―Γ”ΟΒΞ≤®≥ΛΦΛΖΔ”ΪΙβΘ§“ΜΑψ «―Γ‘ώ”ΪΙβΦΛΖΔΙβΤΉ÷–ΒΡΉν¥σΦΛΖΔ≤®≥ΛΉςΈΣΦΛΖΔ≤®≥Λ(4)―Γ‘ώ…®ΟηΖΫ ΫΘΚΗυΨί…®Οη ±Ιβ χΒΡΙλΦΘ≤ΜΆ§Θ§…®ΟηΖΫ Ϋ”–÷± Ϋ…®ΟηΓΔΨβ≥ί…®ΟηΓΔ‘≤–Έ…®ΟηΓΔ«ψ–±…®ΟηΦΑΕύΆ®ΒάΉ‘Ε·…®ΟηΓΘ ΒΦ ΙΛΉς÷–Θ§¥σΕύ≤…”ΟΨβ≥ί…®ΟηΓΘ
…®Οη ±Θ§”ΠΉ‘œ¬Εχ…œ…®ΟηΘ§≤ΜΩ…Καœρ…®ΟηΓΘ…®ΟηΙβ χ(Βψ)ΒΡ–ΈΉ¥ΚΆ¥σ–ΓΘ§Ω…Ά®ΙΐΒς’ϊœΝΖλΒΡΗΏΕ»ΚΆΩμΕ» Βœ÷ΓΘ
ΔΌ÷± Ϋ…®Οη(œΏ–‘…®Οη):“‘“ΜΕ®≤®≥ΛΚΆΩμΕ»ΒΡΙβ χ’’‘Ύ±Γ≤ψΑεΒΡ“ΜΕΥΘ§±Γ≤ψΑεœύΕ‘”ΎΙβ χΉωΒ»ΥΌ÷±œΏ“ΤΕ·÷ΝΝμ“ΜΕΥΓΘ
Ϋχ––÷± Ϋ…®Οη ±Θ§Ιβ χ”Π¬‘Ωμ”ΎΑΏΒψΒΡ÷±ΨΕΘ§ΫΪ’ϊΗωΑΏΒψΑϋά®‘ΎΡΎΘ§≤Δ ΙΑΏΒψ‘ΎΙβ χΒΡ÷––ΡΓΘ”…”ΎΑΏΒψΒΡ÷––Ρ≈®Ε»¥σΘ§±Ώ‘Β≈®Ε»–ΓΘ§Υυ“‘…®ΟηΒΟΒΫΒΡ «Ιβ χ‘ΎΗςΗω≤ΩΖ÷ΒΡΈϋΙβΕ»÷°ΚΆΓΘΗΟΖΫΖ®ΨΏ”–…®ΟηΥΌΕ»ΩλΓΔΉΑ÷ΟΦρΒΞΒΡΧΊΒψΓΘ
»τΑΏΒψ–ΈΉ¥≤ΜΙφ‘ρΘ§‘ρ≤ΜΡή Ι”Ο÷± Ϋ…®ΟηΓΘΕ‘”ΎΆβ–ΈΙφ‘ρΒΡ‘≤–ΈΑΏΒψΓΔΧθΉ¥ΑΏΒψΦΑ”ΪΙβ…®ΟηΖ®ΒΡΑΏΒψΘ§≤…”Ο÷± Ϋ…®Οη±»ΫœΦρΒΞΓΘ
ΔΎΨβ≥ί…®Οη(Ζ…Βψ…®Οη):”Ο“ΜΕ®¥σ–ΓΒΡ’ΐΖΫ–ΈΫΊΟφΜΐΒΡΙβ χ’’…δΑΏΒψΒΡ«ΑΕΥΘ§ΫΪ±Γ≤ψΑεΉωX÷αΒΡΒ»ΥΌ÷±œΏ“ΤΕ·ΒΡΆ§ ±ΉωY÷αΒΡΒ»ΖυΑΎΕ·Θ§ ΙΙβ χΒΡΙλΦΘ≥ Ψβ≥ίΉ¥ΜρΨΊ
–ΈΓΘΉωΨβ≥ί…®Οη ±Θ§”…”ΎΙβ χά¥ΜΊΆ®ΙΐΑΏΒψΘ§“ρ¥ΥΘ§ΑΏΒψΒΡΈϋΙβΕ»ΜΐΖ÷÷Β±»÷± Ϋ…®Οη¥σΓΔ÷Ίœ÷–‘ΚΟΘ§Ρήœϊ≥ΐΖ÷≤ΦΈσ≤νΘ§ ”Ο”ΎΆβ–Έ≤ΜΙφ‘ρΑΏΒψΒΡ…®ΟηΓΘ
“ΜΑψΨβ≥ί…®ΟηΒΡΙβΒψΗΏ1.25 mm,Ωμ1.25 mm,»τΈΣΗΏ–ß±Γ≤ψ…®ΟηΘ§Ω…Βς÷ΝΗΏ0.6mm,Ωμ0.6mmΓΘ
Δέ‘≤–Έ…®ΟηΘΚ”Ο”Ύ‘≤–Ρ ΫΜρœρ–Ρ Ϋ’ΙΩΣΚσΥυΒΟΒΡ‘≤–Έ…ΪΤΉΒΡ…®Οη≤βΕ®ΓΘ
a.ΨΕœρ…®ΟηΘΚΙβ χ”…‘≤–Ρœρ‘≤÷ήΖΫœρΨΕœρ…®ΟηΘ§…®ΟηΙλΦΘΓΘ
b.‘≤÷ή…®ΟηΘΚΙβ χ―Ί“ΜΕ®ΑκΨΕΒΡ‘≤÷ήΖΫœρ“ΤΕ·Θ§Ιβ χ≥Λ÷αΩ…”κ‘≤÷ή“Μ÷¬Θ§“≤Ω…”κ‘≤ ÷ή¥Ι÷±Θ§…®ΟηΙλΦΘΓΘ
Δή«ψ–±…®ΟηΘΚΒ±’ΙΩΣΆ®ΒάΖΔ…ζ«ψ–± ±Θ§…®ΟηΨΆ”Π―ΊΉ≈«ψ–±ΚσΒΡΆ®ΒάΫχ––ΓΘ
ΔίΕύΆ®ΒάΉ‘Ε·…®ΟηΘΚ“«ΤςΧαΙ©ΒΡ≥Χ–ρΖΫ ΫΘ§Ω…Άξ≥…ΕύΆ®ΒάΉ‘Ε·…®ΟηΓΘ–η“Σ…ηΕ®ΤΏ÷÷ ≤Έ ΐΘ§Φ¥≤®≥ΛΠΥsΦΑΠΥRΓΔ…®ΟηΆ®ΒάΒΡXΈΜ÷ΟΓΔΆ®ΒάYΖΫœρΒΡΤπΒψΈΜ÷Ο”κ÷’ΒψΈΜ÷ΟΓΔΆ®Βά ΦδΨύάκ“‘ΦΑ“Μ¥ΈΉ‘Ε·…®ΟηΒΡΆ®Βά ΐΡΩΓΘ“Μ¥ΈΉνΕύΩ……®Οη30ΗωΆ®ΒάΘ§ΟΩ…®ΟηΆξ1ΗωΆ®
ΒάΘ§ΨΆΩ…¥ρ”Γ≥ωΉνΕύ10Ηω…ΪΤΉΖεΒΡΖεΟφΜΐ
6
Κ§ΝΩΦΤΥψ
(1)Άβ±ξ“ΜΒψΖ®ΘΚΒ±±ξΉΦ«ζœΏΆ®Ιΐ‘≠Βψ(ΫΊΨύΈΣΝψ) ±Θ§”ΟΆβ±ξ“ΜΒψΖ®Ε®ΝΩΓΘΦΤΥψ ΙΪ ΫΘΚ
C=F₁ ΓΛA
Ϋ÷–ΘΚCΈΣ±Μ≤βΉιΖ÷ΒΡ≈®Ε»Μρ÷ΊΝΩΘ§AΈΣ±Μ≤βΉιΖ÷ΒΡΖεΟφΜΐΘ§F₁ΈΣ÷±œΏΒΡ–±¬ Μρ±»άΐ≥Θ ΐΓΘ
Ήœ»Ά®ΙΐΕ‘’’ΤΖ»ή“ΚΒΡ≈®Ε»Μρ÷ΊΝΩ”κΕ‘’’ΤΖΒΡΖεΟφΜΐ«σ≥ωF₁,‘ΌΗυΨίΙ© ‘ΤΖΒΡΖεΟφΜΐΚΆF(÷±œΏΒΡ–±¬ Μρ±»άΐ≥Θ ΐ)«σ≥ωΙ© ‘ΤΖΒΡ≈®Ε»Μρ÷ΊΝΩΓΘ
(2)Άβ±ξΝΫΒψΖ®ΘΚΒ±±ξΉΦ«ζœΏ≤ΜΆ®Ιΐ‘≠Βψ ±Θ§”ΟΆβ±ξΝΫΒψΖ®Ε®ΝΩΓΘΦΤΥψΙΪ ΫΘΚ
C=F₁ ΓΛA+F₂
Ϋ÷–ΘΚCΈΣ±Μ≤βΉιΖ÷ΒΡ≈®Ε»Μρ÷ΊΝΩΘ§AΈΣ±Μ≤βΉιΖ÷ΒΡΖεΟφΜΐΘ§F₁ΈΣ÷±œΏΒΡ–±¬ Μρ±»άΐ ≥Θ ΐΘ§F₂ΈΣ÷±œΏ‘ΎΉί÷αΜρΚα÷α…œΒΡΫΊΨύΓΘ
Ήœ»ΗυΨίΕ‘’’ΤΖΒΡΝΫ÷÷≈®Ε»(C₁ΓΔC₂)ΒΡ»ή“ΚΦΑΤδΈϋ ’ΖεΒΡΖεΟφΜΐ(A₁ΓΔA₂)«σ≥ωF₁ΚΆF₂,»ΜΚσάϊ”Οœ¬ ΫΘ§ΗυΨίΙ© ‘ΤΖΒΡΖεΟφΜΐ«σ≥ωΙ© ‘ΤΖ»ή“ΚΒΡ≈®Ε»Μρ÷ΊΝΩΓΘ
7
Φ«¬Φ
Φ«¬Φ“«Τς–ΆΚ≈ΓΔ±Γ≤ψΧθΦΰΓΔ“«Τς≤Έ ΐΓΔœύΙΊ ΐΨίΒ»
8
ΫαΙϊ≈–Ε®
ΫΪ≤βΕ®ΫαΙϊ”κΖ®Ε®“©ΤΖ±ξΉΦœύΙΊΤΖ÷÷Κ§ΝΩ≤βΕ®œνœ¬±ξΉΦΫχ––Ε‘±»Θ§ΖϊΚœ“Σ«σΦ¥ΈΣΚœΗώ
ΚΥ–ΡΡήΝΠ»ΐ Ρή’ΐ»Ζ‘Υ”ΟΗΏ–ß“Κœύ…ΪΤΉΖ®Ϋχ––Κ§ΝΩ≤βΕ®
“ΜΓΔΚΥ–ΡΗ≈Ρν
1.ΗΏ–ß“Κœύ…ΪΤΉΖ® ≤…”ΟΗΏ―Ι δ“Κ±ΟΫΪΝςΕ·œύ±Ο»κΉΑ”–Χν≥δΦΝΒΡ…ΪΤΉ÷υΫχ––Ζ÷άκ≤Δ≤βΕ®Κ§ΝΩΒΡΖΫΖ®ΓΘ
2.…ΪΤΉΝς≥ω«ζœΏΚΆ…ΪΤΉΖε ”…Φλ≤βΤς δ≥ωΒΡ–≈Κ≈«ΩΕ»”κ ±ΦδΉςΆΦΘ§ΥυΒΟΒΡ«ζœΏ≥ΤΈΣ…ΪΤΉΝς≥ω«ζœΏΓΘ«ζœΏ…œΆΜΤπ≤ΩΖ÷ΈΣ…ΪΤΉΖεΓΘ
3.ΜυœΏ ‘Ύ≤ΌΉςΧθΦΰœ¬Θ§…ΪΤΉ÷υ÷–Ϋω”–ΝςΕ·œύΆ®Ιΐ ±Θ§Φλ≤βΤςœλ”Π–≈Κ≈ΒΡΦ«¬Φ÷Β≥ΤΈΣΜυœΏΓΘΈ»Ε®ΒΡΜυœΏ”Π «“ΜΧθ”κΚα÷αΤΫ––ΒΡ÷±œΏΓΘΜυœΏΡήΖ¥”≥“«Τς(÷ς“ΣΈΣΦλ≤βΤς)ΒΡ‘κ…υΥφ ±ΦδΒΡ±δΜ·ΓΘ
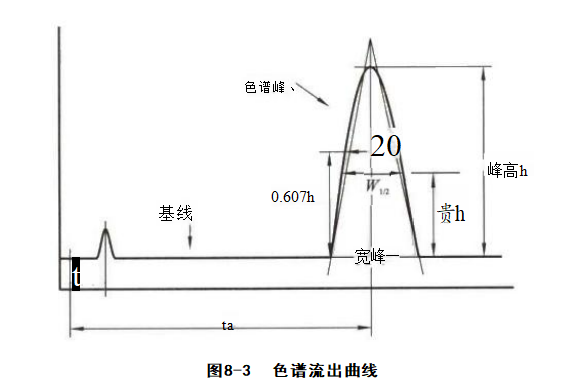
4.ΖεΗΏ(h) …ΪΤΉΖεΕΞΒψ”κΜυœΏΒΡ¥Ι÷±Ψύάκ≥ΤΈΣΖεΗΏ(h),Ω…”Ο”Ύ±Μ≤βΉιΖ÷ΒΡΕ®ΝΩΦΤΥψΓΘ
5.ΖεΟφΜΐ(A) …ΪΤΉΖεΒΡΜΐΖ÷ΟφΜΐ≥ΤΈΣΖεΟφΜΐΘ§Ω…”Ο”Ύ±Μ≤βΉιΖ÷ΒΡΕ®ΝΩΦΤΥψΓΘ
6.±ΘΝτ ±Φδ(tr) ¥”Ϋχ―υΒΫ±Μ≤βΉιΖ÷‘Ύ÷υΚσ≥ωœ÷…ΪΤΉΖεΦΪ¥σ÷Β ±ΥυΨ≠άζΒΡ ±Φδ≥ΤΈΣ±ΘΝτ ±ΦδΓΘ±ΘΝτ ±ΦδΩ…“‘”Ο”Ύ±Μ≤βΉιΖ÷ΒΡΕ®–‘ΓΘ
7.ΑκΖεΩμ(W ₁/2) ΖεΗΏ“ΜΑκ¥ΠΕ‘”ΠΒΡΖεΩμ≥ΤΈΣΑκΖεΩμΘ§W¹/z=2.355Π“ΓΘ
8. ΖεΩμ(W) Ά®Ιΐ…ΪΤΉΖεΝΫ≤ύΒΡΙ’ΒψΉς«–œΏΘ§‘ΎΜυœΏ…œΒΡΫΊΨύ≥ΤΈΣΖεΩμΘ§W=4g=1.699 W¹/2(ΆΦ8-3)ΓΘ
ΕΰΓΔ―ßœΑΡΩ±ξ
Ρή λΝΖ Ι”ΟΗΏ–ß“Κœύ…ΪΤΉΖ®Ϋχ––÷–“©÷ΤΦΝΒΡΚ§ΝΩ≤βΕ®ΓΘ
»ΐΓΔΜυ±Ψ÷Σ Ε
ΗΏ–ß“Κœύ…ΪΤΉΖ®ΨΏ”–Ζ÷άκ–ßΡήΗΏΓΔ―Γ‘ώ–‘ΚΟΓΔΝιΟτΕ»ΗΏΓΔΖ÷ΈωΥΌΕ»ΩλΓΔ ”ΟΖΕΈßΙψ(Ι© ‘ΤΖ≤Μ–ηΤχΜ·Θ§÷Μ–η÷Τ≥…»ή“ΚΦ¥Ω…)ΓΔΝςΕ·œύ―Γ‘ώ–‘¥σΓΔΝς≥ωΉιΖ÷“Ή ’Φ·ΓΔ…ΪΤΉ÷υΩ…Ζ¥Η¥ Ι”ΟΒΡΧΊΒψΘ§Ε‘”ΎΜ”ΖΔ–‘ΒΆΓΔ»»Έ»Ε®–‘≤νΓΔΖ÷Ή”ΝΩ¥σΓΔάκΉ”–ΆΜ·ΚœΈο”»ΈΣ “ΥΓΘΩΤ―ßΦΦ θ‘ΎΗΏ–ß“Κœύ…ΪΤΉΖ®÷–ΒΡ”Π”ΟΘ§ΈΣΗΏ–ß“Κœύ…ΪΤΉΖ®ΩΊ÷Τ÷–“©ΦΑ÷–“©÷ΤΦΝΒΡ÷ ΝΩΧαΙ©ΝΥΩΤ―ßΜυ¥ΓΚΆΙψάΪΒΡ«ΑΨΑΓΘΗΏ–ß“Κœύ
…ΪΤΉΖ®“―≥…ΈΣ÷–“©ΦΑ÷–“©÷ΤΦΝΚ§ΝΩ≤βΕ®Ήν≥Θ”ΟΒΡΖ÷ΈωΖΫΖ®ΓΘ
ΥΡ ΓΔΡήΝΠ―ΒΝΖ
(“Μ)≤ΌΉς”ΟΈο
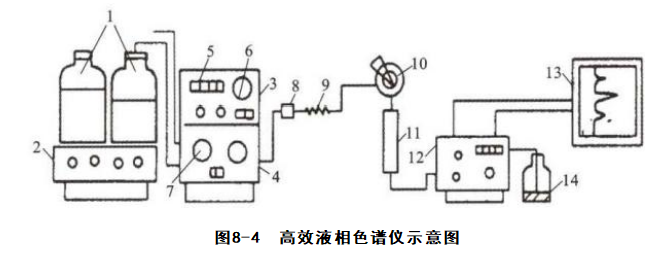
ΗΏ–ß“Κœύ…ΪΤΉ“«“ΜΑψ”…ΗΏ―Ι δ“ΚœΒΆ≥ΓΔΫχ―υœΒΆ≥ΓΔ…ΪΤΉΖ÷άκœΒΆ≥ΓΔΦλ≤βœΒΆ≥ΚΆ ΐΨίΦ«¬Φ”κ¥Π άμœΒΆ≥Ήι≥…ΓΘΥυ”Ο“«Τς”ΠΕ®ΤΎΦλΕ®≤ΔΖϊΚœ”–ΙΊΙφΕ®(ΆΦ8-4ΓΔ±μ8-4)ΓΘ
1.÷ϋ“ΚΙόΘΜ2.Ά―ΤχΉΑ÷ΟΘΜ3.ΧίΕ»œ¥Ά―œΒΆ≥ΘΜ4.ΗΏ―Ι δ“Κ±ΟΘΜ5.ΝςΕ·œύΝςΝΩœ‘ ΨΘΜ6.÷υ«Α―ΙΝΠ±μΘΜ7. δ“Κ±ΟΆΖΘΜ
8.Ιΐ¬ΥΤςΘΜ9.ΉηΡαΤςΘΜ10.Ϋχ―υΉΑ÷ΟΘΜ11.…ΪΤΉ÷υΘΜ12.Φλ≤βΤςΘΜ13. ΐΨί¥ΠάμœΒΆ≥ΘΜ14.Ζœ“Κ÷ϋΙό
±μ8-4 “«ΤςΦρΫι
(Εΰ)ΉΔ“β ¬œν 1.…ΪΤΉ÷υ Ι”ΟΚΆΈ§ΜΛΒΡΉΔ“β ¬œν (1)…ΪΤΉ÷υ”κΫχ―υΤςΦΑΤδ≥ωΩΎΕΥ”κΦλ≤βΤς÷°Φδ”ΠΨΓΝΩΦθ…ΌΥάΧεΜΐΝ§Ϋ”Θ§Φθ…Όά©…ΔΕ‘Ζ÷άκΒΡ”ΑœλΓΘ (2)±ήΟβ―ΙΝΠΦ±Ψγ±δΜ·ΦΑ»ΈΚΈΜζ–Β’ώΕ·ΓΘΩΣΤτ δ“Κ±Ο ±Θ§“Σ÷π≤ΫΦ”¥σ÷ΝΥυ–ηΝςΥΌΘ§±ήΟβΝςΥΌΦ±Ψγ±δΜ·‘λ≥…÷υ―ΙΆΜ»Μ±δ¥σΘ§‘λ≥……ΪΤΉ÷υΙΧΕ®œύΈοάμΥπΜΒΘΜ÷υ―ΙΒΡΆΜ»ΜΫΒΒΆ“≤Μα≥εΕ·÷υΡΎΧνΝœΘ§“ρ¥Υ‘ΎΒςΫΎΝςΥΌ ±”ΠΗΟΜΚ¬ΐΫχ––ΓΘ±ήΟβ…ΪΤΉ÷υ¥”ΗΏ¥ΠΒτœ¬Θ§”Αœλ÷υΡΎΒΡΙΧΕ®œύ(»γ≤ζ…ζΝ―Ζλ)ΓΘ (3)”Π÷πΫΞΗΡ±δ»ήΦΝΒΡΉι≥…Θ§ΧΊ±π «Ζ¥œύ…ΪΤΉ÷–Θ§≤Μ”Π÷±Ϋ”¥””–Μζ»ήΦΝΗΡ±δΈΣ»Ϊ≤Ω «Υ°Θ§Ζ¥÷°“ύ»ΜΓΘ (4)“ΜΑψΕχ―‘Θ§…ΪΤΉ÷υ≤ΜΡήΖ¥≥εΘ§÷Μ”–…ζ≤ζ’Ώ÷ΗΟςΗΟ÷υΩ…“‘Ζ¥≥ε ±Θ§≤≈Ω…“‘Ζ¥≥ε≥ΐ»ΞΝτ‘Ύ÷υΆΖΒΡ‘”÷ ΓΘΖώ‘ρΖ¥≥εΜα―ΗΥΌΫΒΒΆ÷υ–ßΓΘ (5)‘ΎΕ‘–¬÷υΜρ±ΜΈέ»Ψ÷υΫχ––≥εœ¥ ±Θ§”ΠΫΪΤδ≥ωΩΎΕΥ”κΦλ≤βΤςΆ―ΩΣΘ§±ήΟβΈέ»ΨΦλ≤βΤςΓΘ (6)ΗυΨίΝςΕ·œύΒΡ–‘÷ (”»Τδ «pH) ―Γ‘ώ Ι”Ο “ΥΒΡ…ΪΤΉ÷υΘ§“‘±ήΟβΙΧΕ®œύ±ΜΤΤΜΒΓΘ“‘ΙηΫΚΈΣΜυ÷ ΒΡΧνΝœΘ§ΝςΕ·œύΒΡpH”ΠΩΊ÷Τ‘Ύ2~8ΦδΓΘΒ±pH>8 ±Θ§Ω… Ι‘ΊΧεΙηΫΚ»ήΫβΘΜΒ±pH<2 ±Θ§”κΙηΫΚœύΝ§ΒΡΜ·―ßΦϋΚœœύ“ΉΥ°ΫβΆ―¬δΓΘΒ±…ΪΤΉœΒΆ≥÷––η Ι”ΟpH>8ΒΡΝςΕ·œύ ±Θ§”Π―Γ”ΟΡΆ ΦνΧν≥δΦΝΒΡ…ΪΤΉ÷υΘ§Β±–η Ι”ΟpH<2 ΒΡΝςΕ·œύ ±Θ§”Π―Γ”ΟΡΆΥαΧν≥δΦΝΒΡ…ΪΤΉ÷υΓΘ (7)“‘ΙηΫΚΈΣ‘ΊΧεΒΡΦϋΚœΙΧΕ®œύΒΡ Ι”ΟΈ¬Ε»Ά®≥Θ≤Μ≥§Ιΐ40Γφ,ΈΣΗΡ…ΤΖ÷άκ–ßΙϊΘ§Ω… Β±Χα ΗΏ…ΪΤΉ÷υΒΡ Ι”ΟΈ¬Ε»Θ§ΒΪ≤Μ“Υ≥§Ιΐ60ΓφΓΘ»γΙϊ≥§Ιΐ60Γφ,Ω…ΡήΜαΒΦ÷¬…ΪΤΉ÷υΥπΜΒΕχΈόΖ® Ι”ΟΓΘ (8)±ήΟβΫΪΜυ÷ Η¥‘”ΒΡ―υΤΖ”»Τδ «…ζΈο―υΤΖ÷±Ϋ”ΉΔ»κ÷υΡΎΘ§–η“ΣΕ‘―υΤΖΫχ––‘Λ¥ΠάμΜρ’Ώ‘ΎΫχ―υΤςΚΆ…ΪΤΉ÷υ÷°ΦδΝ§Ϋ”“Μ±ΘΜΛ÷υΓΘ±ΘΜΛ÷υ“ΜΑψ «Χν”–œύΥΤΙΧΕ®œύΒΡΕΧ÷υΓΘ±ΘΜΛ÷υΩ…“‘Εχ«“ ”ΠΗΟΨ≠≥ΘΗϋΜΜΓΘ (9)±Θ¥φC18…ΪΤΉ÷υ ±”Π Ι÷υΡΎ≥δ¬ζ““κφΜρΦΉ¥ΦΘ§÷υΫ”ΆΖ“Σ≈ΓΫτΘ§Ζά÷Ι»ήΦΝΜ”ΖΔΗ…‘οΘ§÷υ¥≤ ’ΥθΜρΗ…ΩίΓΘΨχΕ‘Ϋϊ÷ΙΫΪΜΚ≥ε“ΚΝτ‘Ύ÷υΡΎΨ≤÷ΟΙΐ“ΙΜρΗϋ≥Λ ±ΦδΓΘ 2.±ΟΒΡΉΔ“β ¬œν (1)Ζά÷Ι»ΈΚΈΙΧΧεΈΔΝΘΫχ»κ±ΟΧεΘ§“ρΈΣ≥ΨΑΘΜρΤδΥϊ»ΈΚΈ‘”÷ ΈΔΝΘΕΦΜαΡΞΥπ÷υ»ϊΓΔΟήΖβΜΖΓΔΗΉΧεΚΆΒΞœρΖßΘ§“ρ¥Υ”Π‘Λœ»≥ΐ»ΞΝςΕ·œύ÷–ΒΡ»ΈΚΈΙΧΧεΈΔΝΘΓΘΝςΕ·œύ≥ΐ»ΞΩ≈ΝΘΈο÷ ΒΡΖΫΖ® «”Ο0.45ΠΧm ¬ΥΡΛΙΐ¬ΥΓΘ (2)±ΟΒΡ»κΩΎ”ΠΝ§Ϋ”…Α¬ΥΑτ(ΜρΤ§), δ“Κ±ΟΒΡ¬ΥΤς”ΠΨ≠≥Θ«εœ¥ΜρΗϋΜΜΓΘ (3)ΝςΕ·œύ≤Μ”ΠΚ§”–»ΈΚΈΗ· ¥–‘Έο÷ Θ§Κ§”–ΜΚ≥ε“ΚΒΡΝςΕ·œύ≤Μ”Π±ΘΝτ‘Ύ±ΟΡΎΓΘ»γΙϊΫΪΚ§ΜΚ≥ε“ΚΒΡΝςΕ·œύΝτ‘Ύ±ΟΡΎΘ§”…”Ύ’τΖΔΜρ–Ι¬©Θ§…θ÷Ν÷Μ «”…”Ύ»ή“ΚΒΡΨ≤÷ΟΘ§ΨΆΩ…ΡήΈω≥ω―ΈΒΡΈΔœΗΨßΧεΓΘ’β–©ΨßΧεΫΪΥπΜΒΟήΖβΜΖΚΆ÷υ»ϊΒ»ΓΘ (4)±ΟΙΛΉς ±“ΣΖά÷Ι»ήΦΝΤΩΡΎΒΡΝςΕ·œύ±Μ”ΟΆξΘ§Ζώ‘ρΘ§Ω’±Ο‘ΥΉΣΜαΡΞΥπ÷υ»ϊΓΔΗΉΧεΜρΟήΖβΜΖΘ§Ήν÷’ΒΦ÷¬¬©“ΚΓΘ (5) δ“Κ±ΟΒΡΙΛΉς―ΙΝΠΨω≤Μ“Σ≥§ΙΐΙφΕ®ΒΡΉνΗΏ―ΙΝΠΘ§Ζώ‘ρΜα ΙΗΏ―ΙΟήΖβΜΖ±δ–ΈΘ§ΒΦ÷¬¬©“ΚΓΘ 3.ΝυΆ®ΖßΫχ―υΤς Ι”ΟΉΔ“β ¬œν (1) ÷±ζ¥Π”ΎLOADΚΆINJECT÷°Φδ ±Θ§”…”Ύ‘ί ±Ε¬ΉΓΝΥΝς¬ΖΘ§Νς¬Ζ÷–―ΙΝΠ÷η‘ωΘ§‘ΌΉΣΒΫΫχ―υΈΜΘ§ΙΐΗΏΒΡ―ΙΝΠ‘Ύ÷υΆΖ…œ“ΐΤπΥπΜΒΘ§Υυ“‘”ΠΨΓΩλΉΣΕ·ΖßΘ§≤ΜΡήΆΘΝτ‘Ύ÷–ΆΨΓΘ (2)ΗΏ–ß“Κœύ…ΪΤΉ“«Ϋχ―υ Ι”ΟΒΡΉΔ…δΤς’κΆΖ «ΤΫΆΖΉΔ…δΤςΓΘ“ΜΖΫΟφΘ§’κΆΖΆβ≤ύΫτΧυΫχ―υΤςΟήΖβΙήΡΎ≤ύΘ§ΟήΖβ–‘ΡήΚΟΘ§≤Μ¬©“ΚΘ§≤Μ“ΐ»κΩ’ΤχΘΜΝμ“ΜΖΫΟφΘ§“≤Ζά÷ΙΝΥ’κΆΖ¥ΧΜΒΟήΖβΉιΦΰΦΑΕ®Ή”ΓΘ (3)ΝυΆ®ΖßΫχ―υΤςΒΡΫχ―υΖΫ Ϋ”–≤ΩΖ÷ΉΑ“ΚΖ®ΚΆΆξ»ΪΉΑ“ΚΖ®ΝΫ÷÷ΓΘ (4) Ι”ΟΈΔΝΩΉΔ…δΤςΕ®ΝΩ ±Θ§Ϋχ―υΝΩ≤Μ“Υ≥§ΙΐΕ®ΝΩΜΖΧεΜΐΒΡ50%,»γ20ΠΧlΒΡΕ®ΝΩΜΖΉνΕύΫχ10ΠΧlΒΡ―υΤΖΘ§≤Δ«““Σ«σΟΩ¥ΈΫχ―υΧεΜΐΉΦ»ΖΓΔœύΆ§ΘΜ Ι”ΟΕ®ΝΩΜΖΫχ―υ ±Θ§Ϋχ―υΝΩΉν…ΌΈΣΕ®ΝΩΜΖΧεΜΐΒΡ3~5±ΕΘ§Φ¥20ΠΧlΒΡΕ®ΝΩΜΖΉν…ΌΫχ―υ60ΓΪ100ΠΧl ΒΡ―υΤΖΘ§’β―υ≤≈ΡήΆξ»Ϊ÷ΟΜΜ―υΤΖΕ®ΝΩΜΖΡΎ≤–ΝτΒΡ»ή“ΚΘ§¥οΒΫΥυ“Σ«σΒΡΨΪΟήΕ»ΦΑ÷Ίœ÷–‘ΓΘΆΤΦω≤…”Ο100ΠΧl ΒΡΤΫΆΖΫχ―υ’κ≈δΚœ20ΠΧl ΒΡΕ®ΝΩΜΖ¬ζΜΖΫχ―υΓΘ (5)Ϋχ―υ―υΤΖ“Σ«σΈόΈΔΝΘΘ§―υΤΖ»ή“ΚΨυ“Σ”Ο0.45ΠΧm ΒΡ¬ΥΡΛΙΐ¬ΥΓΘ 4.ΤδΥϊΉΔ“β ¬œν (1)≈δ÷ΤΝςΕ·œύ”Π”Ο…ΪΤΉ¥Ω ‘ΦΝΚΆΗΏ¥ΩΥ°ΓΘ”Π”Ο0.45ΠΧm ¬ΥΡΛΙΐ¬Υ≥ΐ»ΞΩ≈ΝΘΈο÷ ΚσΜΙ”ΠΆ―ΤχΘ§“‘Οβ‘Ύ±ΟΡΎ≤ζ…ζΤχ≈ίΘ§”ΑœλΝςΝΩΒΡΈ»Ε®–‘ΘΜ»γΙϊ”–¥σΝΩΤχ≈ίΘ§±ΟΨΆΈόΖ®’ΐ≥ΘΙΛΉςΘΜ»τΤχ≈ίΫχ»κ…ΪΤΉ÷υΚΆΦλ≤βΤςΘ§Μα≥ωœ÷÷υ―Ι≤ΜΈ»ΚΆΜυœΏ≤ΜΈ»Θ§―œ÷Ί ±ΈόΖ®’ΐ≥ΘΦλ≤βΓΘ (2) Ι”ΟΒΡΝςΕ·œύ”ΠΡή”κ“«ΤςœΒΆ≥ΒΡ‘≠±Θ¥φ»ήΦΝΜΞ»ήΘ§»γ≤ΜΜΞ»ήΘ§‘ρœ»»Γœ¬…œ¥ΈΒΡ…ΪΤΉ÷υΘ§”Ο“λ±ϊ¥Φ≥εœ¥ΙΐΕ…Θ§Ϋχ―υΤςΚΆΦλ≤βΤςΒΡΝςΆ®≥Ί“≤ΉΔ»κ“λ±ϊ¥ΦΫχ––ΙΐΕ…Θ§ΙΐΕ…Άξ±œΚσΘ§Ϋ”…œœύ”ΠΒΡ…ΪΤΉ÷υΘ§ΜΜ…œ±Ψ¥Έ Ι”ΟΒΡΝςΕ·œύΘ§‘ΌΫχ––≤ΌΉςΓΘ (3)‘ΎΖ÷ΈωΆξ±œΚσΘ§”Π≥δΖ÷≥εœ¥…ΪΤΉΝς¬ΖœΒΆ≥(¥”±ΟΓΔΫχ―υΤςΓΔ…ΪΤΉ÷υΒΫΦλ≤βΤςΝςΆ®≥Ί),ΧΊ±π «”ΟΙΐΚ§―ΈΝςΕ·œύΒΡΘ§Ηϋ”ΠΉΔ“βœ»”ΟΥ°Θ§‘Ό”ΟΦΉ¥Φ-Υ°≥δΖ÷≥εœ¥ΓΘ»γΖΔœ÷±Ο¬©“ΚΒ»Ϋœ―œ÷ΊΒΡ«ιΩωΘ§”Π«κ”–Ψ≠―ιΒΡΈ§–ό»Υ‘±Ϋχ––Φλ≤ιΓΔΈ§–όΓΘ (4)≤ΌΉςΫα χΚσ”ΠΧν–¥“«Τς Ι”ΟΦ«¬ΦΚΆΗς…ΪΤΉ÷υΒΡ Ι”ΟΦ«¬ΦΘ§≤ΔΦ«¬Φ±Ψ¥Έ≤β ‘“©ΤΖΦΑ÷υ÷–ΒΡ±Θ¥φ»ήΦΝΓΘ
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||